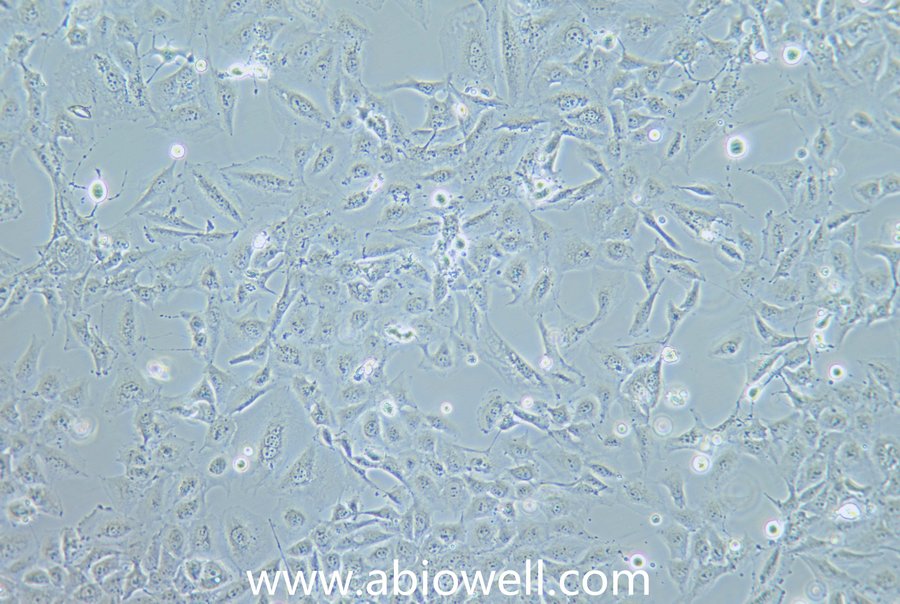
细胞描述:
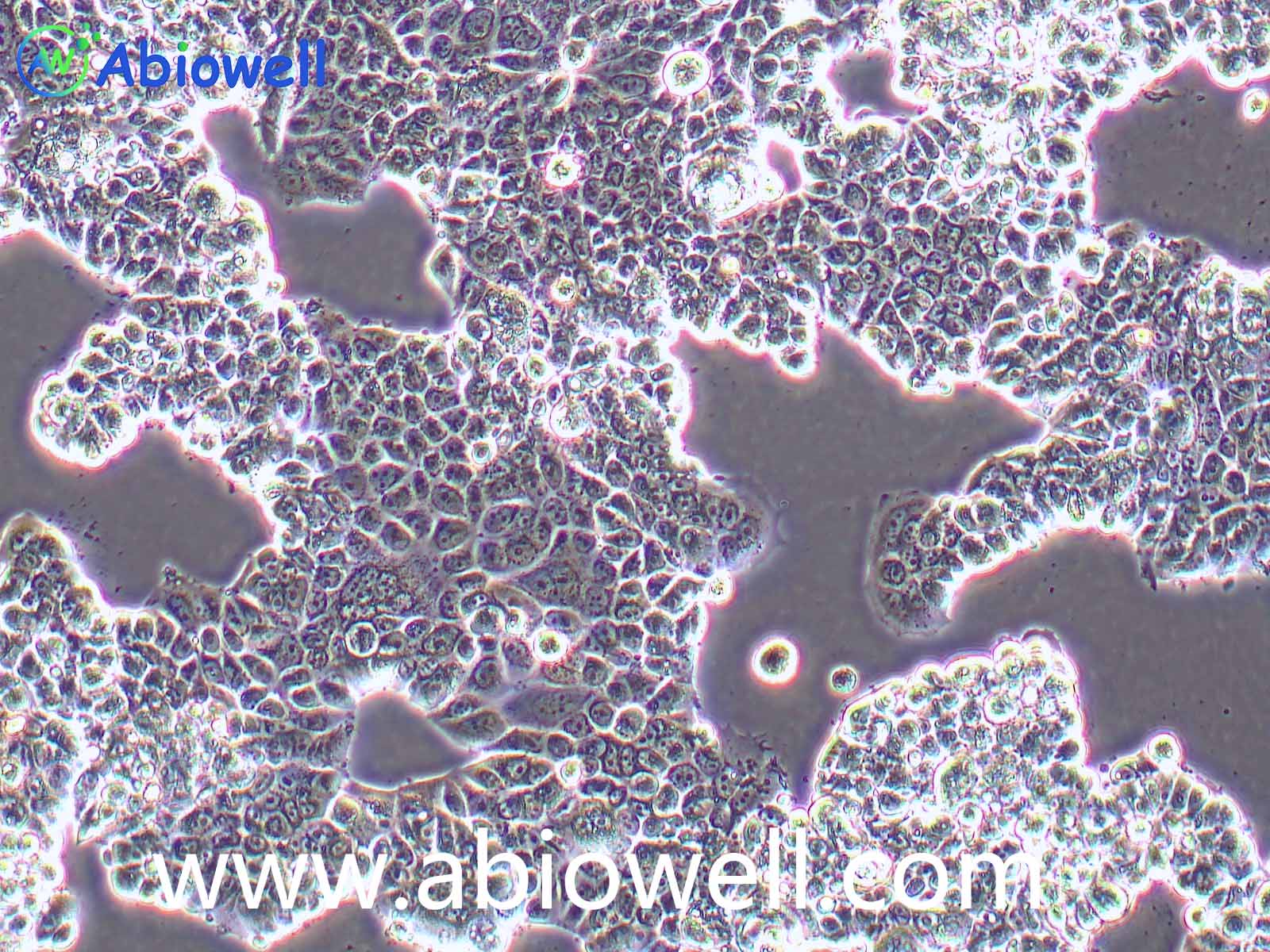
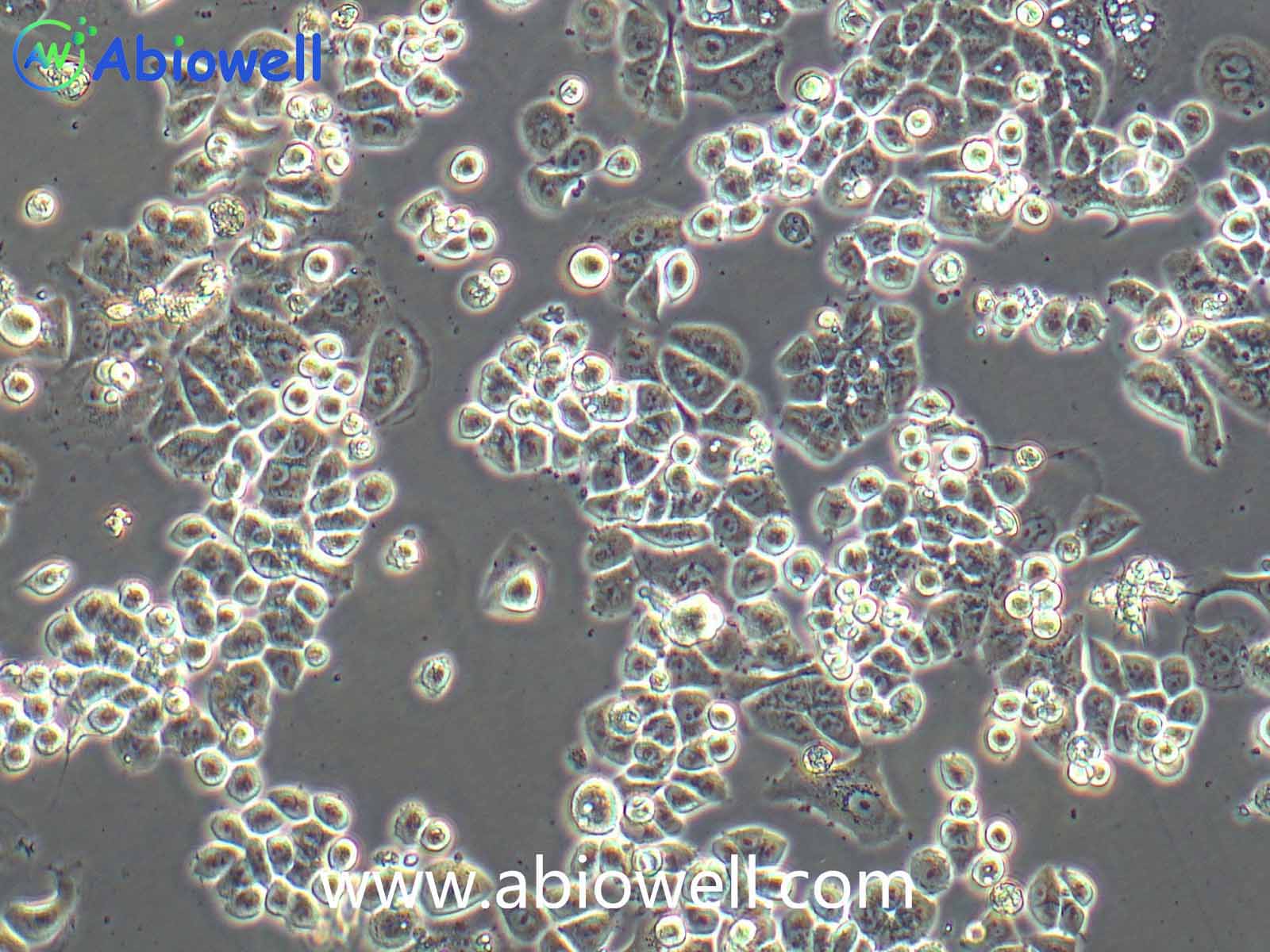
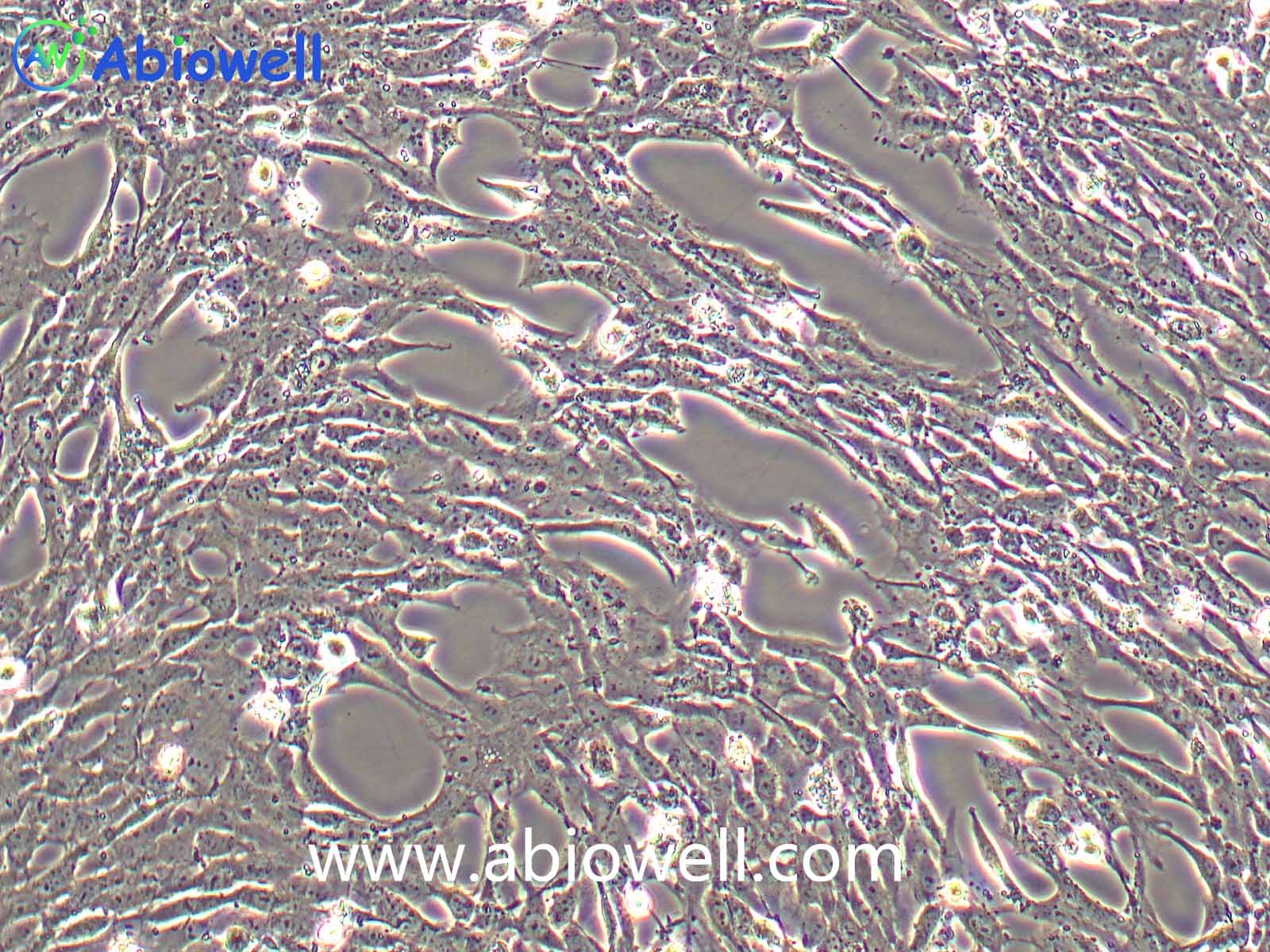
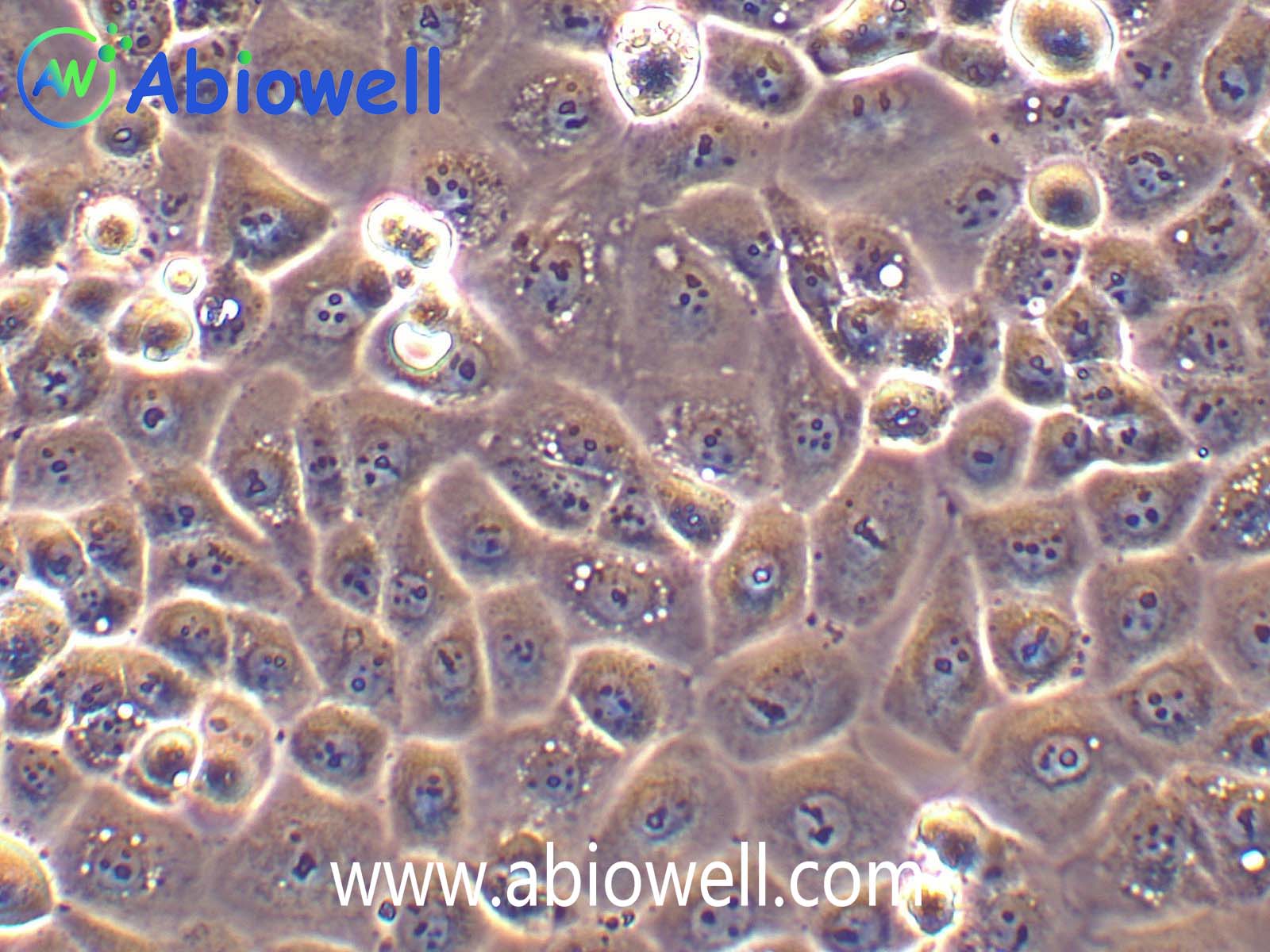
HEK-293T细胞是293T(293tsA1609neo)细胞系(ATCC CRL-11268)的衍生物。细胞持续表达SV40抗原,该细胞常用于转染实验,转染效率较高。(转染一般以50%密度铺板,待细胞刚刚贴到皿壁上后(一般8-10小时),即可进行转染操作)。
细胞特性:
1) 来源:人胚胎肾脏
2) 形态:上皮细胞样,贴壁生长,贴壁能力较弱
3) 规格:1×106cells
4) 培养条件:DMEM+10%FBS+1%P/S (推荐货号AW-MC001)
空气,95%;二氧化碳,5%
37℃
特殊说明:
该细胞贴壁松散,操作时请尽量轻柔;换液时需预热培养基;收货如有大块脱落的细胞团,为正常现象,请按照收货注意事项处理。
细胞接收后的处理:
1) 收到细胞后,活细胞首先观察培养瓶是否完好,培养液是否漏液,培养基是否浑浊;冻存细胞是否干冰已挥发完,冻存管盖是否脱落,破碎,若有这类情况,请务必拍照记录,并于收货24h内与我们联系。
2) 细胞处理:
复苏的细胞:如果是T-25培养瓶活细胞,收到后请用75%的酒精对培养瓶表面进行消毒处理,然后转入培养箱中静置2~3h后再进行后续处理。
备注:运输用的培养基不宜再次用来培养细胞,请按照说明书新配置完全培养基来培养细胞。
冻存细胞:如果是干冰运输的冻存细胞,收到后请立即转入液氮存储或者短暂(24h)放置-80度冰箱保存,或者直接进行细胞复苏。
细胞复苏、传代及冻存流程参考:
1、 细胞复苏
1) 配制完全培养基:基础培养基+胎牛血清+双抗(特殊培养基特殊配置);
2) 细胞复苏:取5ml完全培养基于15ml离心管中,37℃水浴锅预热,从液氮管(或者-80度冰箱)中快速取出冻存的细胞,放入37℃水浴锅中,摇晃使快速化冻(1min左右),然后将化冻的细胞和预热的培养基,移入超净工作台中,化冻的细胞加入到含预热培养基的15ml离心管中,1000rpm离心5min;
3) 吸弃上清,得到细胞沉淀,用2ml完全培养基轻轻重悬细胞,加入到T25培养瓶中,做好标记,放入37℃,5%CO2饱和适度培养箱中培养(培养皿复苏效果更好);
4) 24h后,观察细胞贴壁情况(未贴壁的即为死细胞--针对贴壁细胞),吸弃旧培养基,加入新鲜的预热(室温或37℃)的完全培养基,继续培养。
2、 细胞传代
1) 待细胞生长到80%-90%汇合度时,吸弃旧的培养基,加入1ml无菌PBS润洗一次,以去除残余的培养基及血清(血清含有胰酶的抑制因子),然后加入1ml 0.25%胰酶,37℃培养箱中消化(1~2min左右,不同细胞消化时间不同),取出细胞,镜下观察细胞至细胞皱缩变圆;
2) 加入1ml完全培养基(含FBS)终止消化,轻轻拍打,使细胞脱落下来成单个细胞悬液,收集细胞于15ml无菌离心管中,1000rpm,离心5min;
3) 收集细胞沉淀,完全培养基重悬,一分为二(可根据细胞生长速度调整比例),分别加入到2个新的培养瓶中,做好标记,放入培养箱中培养。
3、细胞冻存
1) 按照细胞传代方法,在超净工作台内消化收集细胞沉淀,取少量细胞用于计数;
2) 用预冷的1ml冻存液(90%完全培养基+10%DMSO)或者无血清细胞冻存液重悬细胞,加入到1.2ml冻存管中,密度为1*106个/ml。
3) 放入程序冻存盒,-80℃过夜后,转入液氮长期保存。
1、ATG8 inhibited endometriosis formation by regulating Treg cells differentiation via integrin α4β1 and Talin-1 interaction
https://www.rbmojournal.com/article/S1472-6483(23)00745-9/abstract